Rasanee Upendra Wanigasuriya1, Asoka Gunaratne2, Aruna Gunapala3
1Senior Registrar in Critical Care Medicine, Colombo South Teaching Hospital, (Sri Lanka)
2Consultant Anesthesiologist, Colombo South Teaching Hospital, (Sri Lanka)
3Consultant Physician, Colombo South Teaching Hospital, (Sri Lanka)
Correspondence: Dr Asoka Gunaratne, Consultant in Anesthetist, Colombo South Teaching Hospital, B229 Hospital Rd, Dehiwala-Mount Lavinia, (Sri Lanka). E-mail: asoka.gunaratne@yahoo.com
ABSTARCT:
Negative pressure pulmonary edema (NPPE) also known as post obstructive pulmonary edema is a potentially life threatening condition with a multifocal pathogenesis. Type 1 NPPE is due to forceful inspiratory effort in the context of an acute airway obstruction, while Type 2 NPPE occurs after relief of a chronic partial upper airway obstruction. Once developed, it impairs gas exchange and causes hypoxemia and if not treated promptly may lead to respiratory failure. The diagnosis of negative pressure pulmonary edema is usually made on the basis of a history of a precipitating incident and symptoms. However, it is basically a diagnosis of exclusion. We present here a case of negative pressure pulmonary edema, which presented as acute left ventricular failure. The cause was eventually found to be acute airway obstruction due to an obstructive goiter precipitated by an upper respiratory tract infection.
Key words: Negative pressure pulmonary edema; Acute left ventricular failure; Goiter
Citation: Wanigasuriya RU, Gunaratne A, Gunapala A. Negative pressure pulmonary edema may present as acute left ventricular failure: a case report. Anaesth Pain & Intensive Care 2017;21(1):98-101
Received: 18 Nov 2016; Reviewed: 12 Dec 2016; Corrected & Accepted: 10 Mar 2017
INTRODUCTION:
Negative pressure pulmonary edema (NPPE) is a form of non-cardiogenic pulmonary edema caused by breathing against an obtruded upper airway. NPPE was first demonstrated in 1927 by Moore in spontaneously breathing dogs exposed to a resistive load.1 The first description of the pathological correlation between creation of negative pressure and the development of pulmonary edema was in 1942 by Warren at al. The relationship between pulmonary edema and upper airway obstruction in two children, who had croup and epiglottitis, was reported in 1973 by Capitanio et al.2 The report by Oswalt et al in 19773 was the first showing the clinical significance of this phenomenon in three adult patients, who experienced the onset of pulmonary edema minutes after severe acute upper airway obstruction. Since then, NPPE has been reported mainly by anesthetists as a consequence of postoperative laryngospasm.4,5
NPPE associated with goiter was reported in the Canadian Medical Association Journal, in a fifty years old women.6 We encountered a rare case of negative pressure pulmonary edema due to an obstructive goiter, which presented as acute left ventricular failure.
CASE REPORT:
A 55 year old female presented with sudden onset shortness of breath and associated expectoration of frothy blood stained sputum. There was no associated chest pain, cough or fever. She had a past history of recent onset exertional dyspnea. She was a diagnosed patient with hypertension and type II diabetes mellitus for the past eight years, with regular clinic follow up. There was no other significant history, except for a partial thyroidectomy done 20 years ago for a multinodular goiter for which she was not on thyroxine replacement.
On admission to Colombo South Teaching Hospital, patient was in respiratory distress and had clinical features of acute pulmonary edema. She was treated with oxygen, continuous positive airway pressure (CPAP) and loop diuretics in the ward. Eventually patient had become hemodynamically unstable; therefore she was intubated and started on an inotrope. Patient was admitted to the intensive care unit for mechanical ventilation and supportive care. On admission patient developed a narrow complex tachycardia with low blood pressure for which DC cardioversion was done twice and converted to sinus rhythm. Afterwards amiodarone infusion was started. There were no ischemic changes in the ECG but due to the given history a tentative diagnosis of acute left ventricular failure following acute coronary syndrome was made and antiplatelets and enoxaparin were started. Chest x-ray showed bilateral hilar congestion suggestive of pulmonary edema. However, a bedside transthoracic echo done subsequently revealed normal LV function with EF – 60%. No regional wall motion abnormalities or pericardial effusion were noted. Troponin level was initially normal but subsequent repeat test after 6 hours was mildly elevated. The brain natriuretic peptide (BNP) level was marginally elevated and the D dimers were negative.
Patient’s hemodynamic parameters gradually improved with support. She was gradually weaned off from ventilator supports and spontaneous breathing trials became successful. Therefore patient was extubated. However within 30 minutes she had to be reintubated due to respiratory distress. IV dexamethasone and adrenalin nebulization was commenced and an extubation was planned on the following day. She was maintaining respiratory parameters for about 6 hours following extubation when she became dyspneic, tachypneic and developed an inspiratory stridor. SpO2 was decreasing therefore she was reintubated. There were widespread bilateral crepitations and ultrasound of lung revealed multiple B lines which suggested fluid in the lung parenchyma. She was given high positive end expiratory pressure (PEEP) and a bolus of frusemide. Chest x-ray taken two hours later was clear and the repeat lung ultrasound showed normal lung. Repeat echo-cardiogram revealed normal cardiac functions.
A clinical diagnosis of negative pressure pulmonary edema due to airway obstruction was suspected. To identify the cause for it, a neck ultrasound was done first. It showed a multinodular goiter with mid tracheal compression. Fibreoptic laryngoscopy was planned by the ENT team. For the procedure patient was extubated for the third time. It revealed normal vocal cords, subglottic edema and inflammatory changes. No significant narrowing was noted. She was stable for about 12 hours when she became distressed and reintubated again. A contrast CT of neck and chest showed diffuse enlargement of the thyroid gland with gross tracheal compression at C7/T1 level measuring a transverse diameter of 3mm.(Figure 1) Thyroidectomy was planned. Patient was kept intubated till the day of surgery.
Total thyroidectomy was performed by the ENT team. Trachea was found to be weak. Strap muscle flap was performed to strengthen the weak trachea. Patient was extubated on postoperative second day. There were no complications afterwards and patient was discharged home on replacement therapy with oral thyroxine and calcium.
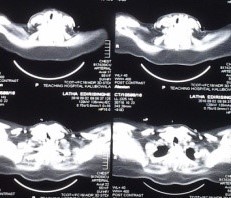
Figure 1: CT Neck showing tracheal compression at C7/T1 level
DISCUSSION:
Negative pressure pulmonary edema also known as post obstructive pulmonary edema7 is a potentially fatal condition with a multifactorial pathogenesis.8 Literature reveals a morbidity and mortality rate of 11% to 40% in reported cases8.
Two distinct subclasses of negative pressure pulmonary edema have been described.7,8 Type I NPPE is associated with forceful inspiratory effort in the context of an acute airway obstruction. Common etiologies are laryngospasm, epiglottitis, croup, endotracheal tube obstruction, strangulation, hanging, goiter and postoperative vocal cord paralysis. In adults the commonest cause is laryngospasms, which has an incidence of approximately 50%.10 Type II NPPE occurs after relief of a chronic partial upper airway obstruction, as maybe expected after adenoidectomy/ tonsillectomy, and laryngeal mass resection.7 In this review, we report a patient who developed Type I NPPE probably following acute airway obstruction due to goiter.
Two different mechanisms have been postulated to explain the pathogenesis of Type I NPPE.11,12 According to the hydrostatic mechanism, a forceful inspiration against an obstructed airway creates a large negative inspiratory pressure. This negative pressure increases the venous return to the heart, thereby increasing the pulmonary venous pressure. It also decreases perivascular interstitial hydrostatic pressure. Therefore, this leads to movement of fluid from the capillaries into the interstitium and alveolar spaces via a hydrostatic pressure gradient resulting in transudative pulmonary edema.12 The mechanical stress mechanism explains that during inspiration against an obstructed upper airway creates a large transmural pulmonary capillary pressure which disrupts the alveolar epithelium and pulmonary microvascular membranes. This leads to increased pulmonary capillary permeability and exudative pulmonary edema.12
Although negative inspiratory pressure is the primary pathophysiology, hypoxia, hypercarbia, acidosis, and hyperadrenergic state all contribute to its development.13
Our patient had an undetected enlarged thyroid gland which caused compression and narrowing of the trachea which led to an obstructed airway. The cause for the sudden complete obstruction maybe due to airway edema due to an undetected respiratory tract infection or hyperventilation leading to collapse of airway due to the venturi effect. The generation of a large negative inspiratory pressure and disruption of the alveolar capillary membrane could have been the underlying pathophysiology for generation of pulmonary edema.
The mechanism behind Type II NPPE is less clear.12 It is postulated that expiratory efforts against a partial chronic obstruction creates an auto PEEP leading to increased intrathoracic pressure, and prevents venous return.14 After relief of obstruction there is a subsequent ‘suction’ of fluid into the pulmonary vasculature leading to transudative pulmonary edema. This mechanism is unlikely in our patient as there was no mechanism such as surgery, to release the obstruction.
The onset of pulmonary edema varies from few minutes to several hours (up to 30 hours) following airway obstruction or sudden release of a chronic obstruction12. The clinical features are tachypnea, tachycardia, pink frothy respiratory secretions, progressive desaturation and crackles on chest auscultation8. Our patient had a similar presentation on admission, even though there was no history to suggest long standing obstruction.
Diagnosis of NPPE is usually made on the basis of a history of a precipitating incident and symptoms.8 However, it is basically a clinical entity which can be established by diagnosis of exclusion.12 The main differential diagnosis includes cardiogenic pulmonary edema, aspiration pneumonia, pulmonary embolism and anaphylaxis.7 Chest radiography usually shows bilateral changes consistent with pulmonary edema in both cardiogenic and negative pressure pulmonary edema. Abnormal BNP, elevated troponin levels and abnormal echocardiogram may suggest cardiogenic pulmonary edema. Elevated D dimers and positive CT pulmonary angiogram (CTPA) indicates pulmonary embolism.
The clinical presentation of our patient was in accordance with acute pulmonary edema. The origin was thought to be cardiogenic as she had a history of long standing diabetes mellitus. In such ischemic heart disease is a high possibility. Even though on admission there were no ischemic changes in the ECG, she had a supraventricular tachycardia which could have been ischemic in origin. Her repeat troponin level and BNP level were marginally elevated. This can be attributed to cardiogenic pulmonary edema. The point against this diagnosis was that the bedside echocardiogram on admission and the repeat echo done later following extubation were normal. There were no clinical features to suggest deep vein thrombosis and the D dimers and bedside echocardiogram were normal. Therefore acute pulmonary embolism was probably unlikely. She had no history to suggest aspiration or anaphylaxis. NPPE was lower down in the differentials because she did not have a history to suggest airway obstruction and the goiter was not prominent as her neck was thick and the patient obese. Therefore a working diagnosis of cardiogenic pulmonary edema was made initially.
However, the patient developed repeated extubation failure, with possible recurrent episodes of pulmonary edema. Recurrent episodes of pulmonary edema is a rare phenomenon with not many reported cases.15 We revised our probable diagnosis to NPPE and investigated for a possible cause, which was later found to be an obstructive goiter.
In diagnosed patients with NPPE, therapy is aimed at reversing hypoxia and removal of excess fluid from lung interstitium.12 Frusemide is often used to promote diuresis and to remove excess intrapulmonary fluid.12 However, diuretics may exacerbate hypovolemia and hypoperfusion, therefore their role in NPPE remains uncertain.13 Non-invasive ventilation has great benefit in reducing the pulmonary congestion. It helps recruitment of alveoli and reduces the work of breathing. In severe cases, patients require mechanical ventilation with PEEP.8
Our patient was treated with oxygen and non-invasive ventilation initially in the ward. Later she was intubated due to respiratory failure and was mechanically ventilated with PEEP until her symptoms resolved.
The resolution of pulmonary edema usually occurs in 3-12 hours of institution of appropriate therapy. However, in few cases it may take up to 48 hours. Patients have a good prognosis if promptly diagnosed and appropriately treated.12 Resolution of pulmonary edema in our patient was slower as her symptoms were severe leading to respiratory failure.
CONCLUSION:
Negative pressure pulmonary edema is a potentially fatal condition with a multifactorial pathogenesis. Its diagnosis will be difficult if an obvious cause of airway obstruction is not present. Rapid recognition and initiation of appropriate therapy reduces the complications and mortality.
Conflict of interest: None declared by the authors
Author contribution: All of the authors took part in management of the case and the manuscript preparation
REFERENCES:
1Senior Registrar in Critical Care Medicine, Colombo South Teaching Hospital, (Sri Lanka)
2Consultant Anesthesiologist, Colombo South Teaching Hospital, (Sri Lanka)
3Consultant Physician, Colombo South Teaching Hospital, (Sri Lanka)
Correspondence: Dr Asoka Gunaratne, Consultant in Anesthetist, Colombo South Teaching Hospital, B229 Hospital Rd, Dehiwala-Mount Lavinia, (Sri Lanka). E-mail: asoka.gunaratne@yahoo.com
ABSTARCT:
Negative pressure pulmonary edema (NPPE) also known as post obstructive pulmonary edema is a potentially life threatening condition with a multifocal pathogenesis. Type 1 NPPE is due to forceful inspiratory effort in the context of an acute airway obstruction, while Type 2 NPPE occurs after relief of a chronic partial upper airway obstruction. Once developed, it impairs gas exchange and causes hypoxemia and if not treated promptly may lead to respiratory failure. The diagnosis of negative pressure pulmonary edema is usually made on the basis of a history of a precipitating incident and symptoms. However, it is basically a diagnosis of exclusion. We present here a case of negative pressure pulmonary edema, which presented as acute left ventricular failure. The cause was eventually found to be acute airway obstruction due to an obstructive goiter precipitated by an upper respiratory tract infection.
Key words: Negative pressure pulmonary edema; Acute left ventricular failure; Goiter
Citation: Wanigasuriya RU, Gunaratne A, Gunapala A. Negative pressure pulmonary edema may present as acute left ventricular failure: a case report. Anaesth Pain & Intensive Care 2017;21(1):98-101
Received: 18 Nov 2016; Reviewed: 12 Dec 2016; Corrected & Accepted: 10 Mar 2017
INTRODUCTION:
Negative pressure pulmonary edema (NPPE) is a form of non-cardiogenic pulmonary edema caused by breathing against an obtruded upper airway. NPPE was first demonstrated in 1927 by Moore in spontaneously breathing dogs exposed to a resistive load.1 The first description of the pathological correlation between creation of negative pressure and the development of pulmonary edema was in 1942 by Warren at al. The relationship between pulmonary edema and upper airway obstruction in two children, who had croup and epiglottitis, was reported in 1973 by Capitanio et al.2 The report by Oswalt et al in 19773 was the first showing the clinical significance of this phenomenon in three adult patients, who experienced the onset of pulmonary edema minutes after severe acute upper airway obstruction. Since then, NPPE has been reported mainly by anesthetists as a consequence of postoperative laryngospasm.4,5
NPPE associated with goiter was reported in the Canadian Medical Association Journal, in a fifty years old women.6 We encountered a rare case of negative pressure pulmonary edema due to an obstructive goiter, which presented as acute left ventricular failure.
CASE REPORT:
A 55 year old female presented with sudden onset shortness of breath and associated expectoration of frothy blood stained sputum. There was no associated chest pain, cough or fever. She had a past history of recent onset exertional dyspnea. She was a diagnosed patient with hypertension and type II diabetes mellitus for the past eight years, with regular clinic follow up. There was no other significant history, except for a partial thyroidectomy done 20 years ago for a multinodular goiter for which she was not on thyroxine replacement.
On admission to Colombo South Teaching Hospital, patient was in respiratory distress and had clinical features of acute pulmonary edema. She was treated with oxygen, continuous positive airway pressure (CPAP) and loop diuretics in the ward. Eventually patient had become hemodynamically unstable; therefore she was intubated and started on an inotrope. Patient was admitted to the intensive care unit for mechanical ventilation and supportive care. On admission patient developed a narrow complex tachycardia with low blood pressure for which DC cardioversion was done twice and converted to sinus rhythm. Afterwards amiodarone infusion was started. There were no ischemic changes in the ECG but due to the given history a tentative diagnosis of acute left ventricular failure following acute coronary syndrome was made and antiplatelets and enoxaparin were started. Chest x-ray showed bilateral hilar congestion suggestive of pulmonary edema. However, a bedside transthoracic echo done subsequently revealed normal LV function with EF – 60%. No regional wall motion abnormalities or pericardial effusion were noted. Troponin level was initially normal but subsequent repeat test after 6 hours was mildly elevated. The brain natriuretic peptide (BNP) level was marginally elevated and the D dimers were negative.
Patient’s hemodynamic parameters gradually improved with support. She was gradually weaned off from ventilator supports and spontaneous breathing trials became successful. Therefore patient was extubated. However within 30 minutes she had to be reintubated due to respiratory distress. IV dexamethasone and adrenalin nebulization was commenced and an extubation was planned on the following day. She was maintaining respiratory parameters for about 6 hours following extubation when she became dyspneic, tachypneic and developed an inspiratory stridor. SpO2 was decreasing therefore she was reintubated. There were widespread bilateral crepitations and ultrasound of lung revealed multiple B lines which suggested fluid in the lung parenchyma. She was given high positive end expiratory pressure (PEEP) and a bolus of frusemide. Chest x-ray taken two hours later was clear and the repeat lung ultrasound showed normal lung. Repeat echo-cardiogram revealed normal cardiac functions.
A clinical diagnosis of negative pressure pulmonary edema due to airway obstruction was suspected. To identify the cause for it, a neck ultrasound was done first. It showed a multinodular goiter with mid tracheal compression. Fibreoptic laryngoscopy was planned by the ENT team. For the procedure patient was extubated for the third time. It revealed normal vocal cords, subglottic edema and inflammatory changes. No significant narrowing was noted. She was stable for about 12 hours when she became distressed and reintubated again. A contrast CT of neck and chest showed diffuse enlargement of the thyroid gland with gross tracheal compression at C7/T1 level measuring a transverse diameter of 3mm.(Figure 1) Thyroidectomy was planned. Patient was kept intubated till the day of surgery.
Total thyroidectomy was performed by the ENT team. Trachea was found to be weak. Strap muscle flap was performed to strengthen the weak trachea. Patient was extubated on postoperative second day. There were no complications afterwards and patient was discharged home on replacement therapy with oral thyroxine and calcium.
Figure 1: CT Neck showing tracheal compression at C7/T1 level
DISCUSSION:
Negative pressure pulmonary edema also known as post obstructive pulmonary edema7 is a potentially fatal condition with a multifactorial pathogenesis.8 Literature reveals a morbidity and mortality rate of 11% to 40% in reported cases8.
Two distinct subclasses of negative pressure pulmonary edema have been described.7,8 Type I NPPE is associated with forceful inspiratory effort in the context of an acute airway obstruction. Common etiologies are laryngospasm, epiglottitis, croup, endotracheal tube obstruction, strangulation, hanging, goiter and postoperative vocal cord paralysis. In adults the commonest cause is laryngospasms, which has an incidence of approximately 50%.10 Type II NPPE occurs after relief of a chronic partial upper airway obstruction, as maybe expected after adenoidectomy/ tonsillectomy, and laryngeal mass resection.7 In this review, we report a patient who developed Type I NPPE probably following acute airway obstruction due to goiter.
Two different mechanisms have been postulated to explain the pathogenesis of Type I NPPE.11,12 According to the hydrostatic mechanism, a forceful inspiration against an obstructed airway creates a large negative inspiratory pressure. This negative pressure increases the venous return to the heart, thereby increasing the pulmonary venous pressure. It also decreases perivascular interstitial hydrostatic pressure. Therefore, this leads to movement of fluid from the capillaries into the interstitium and alveolar spaces via a hydrostatic pressure gradient resulting in transudative pulmonary edema.12 The mechanical stress mechanism explains that during inspiration against an obstructed upper airway creates a large transmural pulmonary capillary pressure which disrupts the alveolar epithelium and pulmonary microvascular membranes. This leads to increased pulmonary capillary permeability and exudative pulmonary edema.12
Although negative inspiratory pressure is the primary pathophysiology, hypoxia, hypercarbia, acidosis, and hyperadrenergic state all contribute to its development.13
Our patient had an undetected enlarged thyroid gland which caused compression and narrowing of the trachea which led to an obstructed airway. The cause for the sudden complete obstruction maybe due to airway edema due to an undetected respiratory tract infection or hyperventilation leading to collapse of airway due to the venturi effect. The generation of a large negative inspiratory pressure and disruption of the alveolar capillary membrane could have been the underlying pathophysiology for generation of pulmonary edema.
The mechanism behind Type II NPPE is less clear.12 It is postulated that expiratory efforts against a partial chronic obstruction creates an auto PEEP leading to increased intrathoracic pressure, and prevents venous return.14 After relief of obstruction there is a subsequent ‘suction’ of fluid into the pulmonary vasculature leading to transudative pulmonary edema. This mechanism is unlikely in our patient as there was no mechanism such as surgery, to release the obstruction.
The onset of pulmonary edema varies from few minutes to several hours (up to 30 hours) following airway obstruction or sudden release of a chronic obstruction12. The clinical features are tachypnea, tachycardia, pink frothy respiratory secretions, progressive desaturation and crackles on chest auscultation8. Our patient had a similar presentation on admission, even though there was no history to suggest long standing obstruction.
Diagnosis of NPPE is usually made on the basis of a history of a precipitating incident and symptoms.8 However, it is basically a clinical entity which can be established by diagnosis of exclusion.12 The main differential diagnosis includes cardiogenic pulmonary edema, aspiration pneumonia, pulmonary embolism and anaphylaxis.7 Chest radiography usually shows bilateral changes consistent with pulmonary edema in both cardiogenic and negative pressure pulmonary edema. Abnormal BNP, elevated troponin levels and abnormal echocardiogram may suggest cardiogenic pulmonary edema. Elevated D dimers and positive CT pulmonary angiogram (CTPA) indicates pulmonary embolism.
The clinical presentation of our patient was in accordance with acute pulmonary edema. The origin was thought to be cardiogenic as she had a history of long standing diabetes mellitus. In such ischemic heart disease is a high possibility. Even though on admission there were no ischemic changes in the ECG, she had a supraventricular tachycardia which could have been ischemic in origin. Her repeat troponin level and BNP level were marginally elevated. This can be attributed to cardiogenic pulmonary edema. The point against this diagnosis was that the bedside echocardiogram on admission and the repeat echo done later following extubation were normal. There were no clinical features to suggest deep vein thrombosis and the D dimers and bedside echocardiogram were normal. Therefore acute pulmonary embolism was probably unlikely. She had no history to suggest aspiration or anaphylaxis. NPPE was lower down in the differentials because she did not have a history to suggest airway obstruction and the goiter was not prominent as her neck was thick and the patient obese. Therefore a working diagnosis of cardiogenic pulmonary edema was made initially.
However, the patient developed repeated extubation failure, with possible recurrent episodes of pulmonary edema. Recurrent episodes of pulmonary edema is a rare phenomenon with not many reported cases.15 We revised our probable diagnosis to NPPE and investigated for a possible cause, which was later found to be an obstructive goiter.
In diagnosed patients with NPPE, therapy is aimed at reversing hypoxia and removal of excess fluid from lung interstitium.12 Frusemide is often used to promote diuresis and to remove excess intrapulmonary fluid.12 However, diuretics may exacerbate hypovolemia and hypoperfusion, therefore their role in NPPE remains uncertain.13 Non-invasive ventilation has great benefit in reducing the pulmonary congestion. It helps recruitment of alveoli and reduces the work of breathing. In severe cases, patients require mechanical ventilation with PEEP.8
Our patient was treated with oxygen and non-invasive ventilation initially in the ward. Later she was intubated due to respiratory failure and was mechanically ventilated with PEEP until her symptoms resolved.
The resolution of pulmonary edema usually occurs in 3-12 hours of institution of appropriate therapy. However, in few cases it may take up to 48 hours. Patients have a good prognosis if promptly diagnosed and appropriately treated.12 Resolution of pulmonary edema in our patient was slower as her symptoms were severe leading to respiratory failure.
CONCLUSION:
Negative pressure pulmonary edema is a potentially fatal condition with a multifactorial pathogenesis. Its diagnosis will be difficult if an obvious cause of airway obstruction is not present. Rapid recognition and initiation of appropriate therapy reduces the complications and mortality.
Conflict of interest: None declared by the authors
Author contribution: All of the authors took part in management of the case and the manuscript preparation
REFERENCES:
- Moore RL, Binger CA. The response to repiratory resistance: A comparison of the effects produced by partial obstruction in the inspiratory and expiratory phases of respiration. J Exp Med. 1927 May 31;45(6):1065-80. [PubMed] [Free full text]
- Capitanio MA, Kirkpatrick JA. Obstruction of the upper airway in children as reflected on the chest radiograph. Radiology. 1973 Apr;107(1):159-61. [PubMed]
- Oswalt CE, Gates GA, Holmstrom MG. Pulmonary oedema as a complication of acute airway obstruction. JAMA. 1977 Oct 24;238(17):1833-35. [PubMed]
- Cascade PN, Alexander GD, Mackie DS. Negative pressure pulmonary oedema after endotracheal intubation. Radiology. 1993 Mar;186(3):671-5. [PubMed]
- Dolinski SY, MacGregor DA, Scuderi PE. Pulmonary haemorrhage associated with negative pressure pulmonary oedema. Anaesthesilogy. 2000 Sep;93(3):888-90. [PubMed] [Free full text]
- Liu PY, Shih ML, Chen CW. Post obstructive pulmonary oedema associated with a sub sternal goitre. CMAJ. 2012 Dec 11;184(18):2011-13. doi: 10.1503/cmaj.120256. [PubMed] [Free full text]
- Udeshi A, Cantie SM, Pierre E. Postobstructive pulmonary oedema. J Crit Care. 2010 Sep;25(3):508.e1-5. doi: 10.1016/j.jcrc.2009.12.014 [PubMed] [Free full text]
- Bhaskar B, Fraser JF. Negative pressure pulmonary oedema revisited: pathophysiology and review of management. Saudi J Anaesth. 2011 Jul;5(3):308-13. doi: 10.4103/1658-354X.84108. [PubMed] [Free full text]
- Vishvanathan T, Kluger MT, Webb RK, Westhorpe RN. Crisis management during anaesthesia: laryngospasm. Qual Saf Health Care. 2005 Jun;14(3):e3. [PubMed] [Free full text]
- Louis PJ, Fernandes R. Negative pressure pulmonary oedema. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002 Jan;93(1):4-6. [PubMed]
- Lemyze M, Mallat J. Understanding negative pressure pulmonary oedema. Intensive Care Med. 2014 Aug;40(8):1140-1143. doi: 10.1007/s00134-014-3307-7 [PubMed] [Free full text]
- Ren-Guang Wu. The clinical characteristics of negative pressure pulmonary oedema. J Intern Med Taiwan (2015) 26;63-68. [PubMed] [Free full text]
- McConkey PP. Post obstructive pulmonary oedema: A case series and review. Anaesth Intensive Care. 2000 Feb;28(1):72-6. [PubMed]
- Blanch L, Barnabe F, Lucangelo U. Measurement of air-trapping, intrinsic positive end expiratory pressure and dynamic hyperinflation in mechanically ventilated patients. Respir Care. 2005 Jan;50(1):110-23. [PubMed] [Free full text]
- Pathak V, Rendon IS, Ciubotaru RL. Recurrent negative pressure pulmonary oedema. Clin Med Res. 2011 Jun;9(2): 88–91. doi: 10.3121/cmr.2010.936. [PubMed] [Free full text]